Development of the Reproductive Organs
The development of these reproductive organs begins at an early stage in the embryo.
There is a close link throughout with the development of the urinary system.
The Gonads
Indifferent Stage
In the first stage of gonadal development, it is impossible to distinguish between the male and female gonad. Thus, it is known as the indifferent stage.
The gonads begin as genital ridges – a pair of longitudinal ridges derived from intermediate mesoderm and overlying epithelium. They initially do not contain any germ cells.
In the fourth week, germ cells begin to migrate from the endoderm lining of the yolk sac to the genital ridges, via the dorsal mesentery of the hindgut. They reach the genital ridges in the sixth week.
Simultaneously, the epithelium of the genital ridges proliferates and penetrates the intermediate mesoderm to form the primitive sex cords. The combination of germ cells and primitive sex cords forms the indifferent gonad – from which development into the testes or ovaries can occur.
Testes
In a male embryo, the XY sex chromosomes are present. The Y chromosome contains the SRY gene, which stimulates the development of the primitive sex cords to form testis (medullary) cords. The tunica albuginea, a fibrous connective tissue layer, forms around the cords.
A portion of the testis cords breaks off to form the future rete testis. The remaining cords contain two types of cells:
- Germ cells
- Sertoli cells (derived from the surface epithelium of the gland).
In puberty, these cords acquire a lumen and become the seminiferous tubules – the site within which sperm will be formed.
Located between the testis cords are the Leydig cells (derived from the intermediate mesoderm). In the eighth week, they begin production of testosterone – which drives differentiation of the internal and external genitalia.
Ovaries
In a female embryo, the XX sex chromosomes are present. As there is no Y chromosome, there is no SRY gene to influence development. Without it, the primitive sex cords degenerate and do not form the testis cords.
Instead, the epithelium of the gonad continues to proliferate, producing cortical cords. In the third month, these cords break up into clusters, surrounding each oogonium (germ cell) with a layer of epithelial follicular cells, forming a primordial follicle.
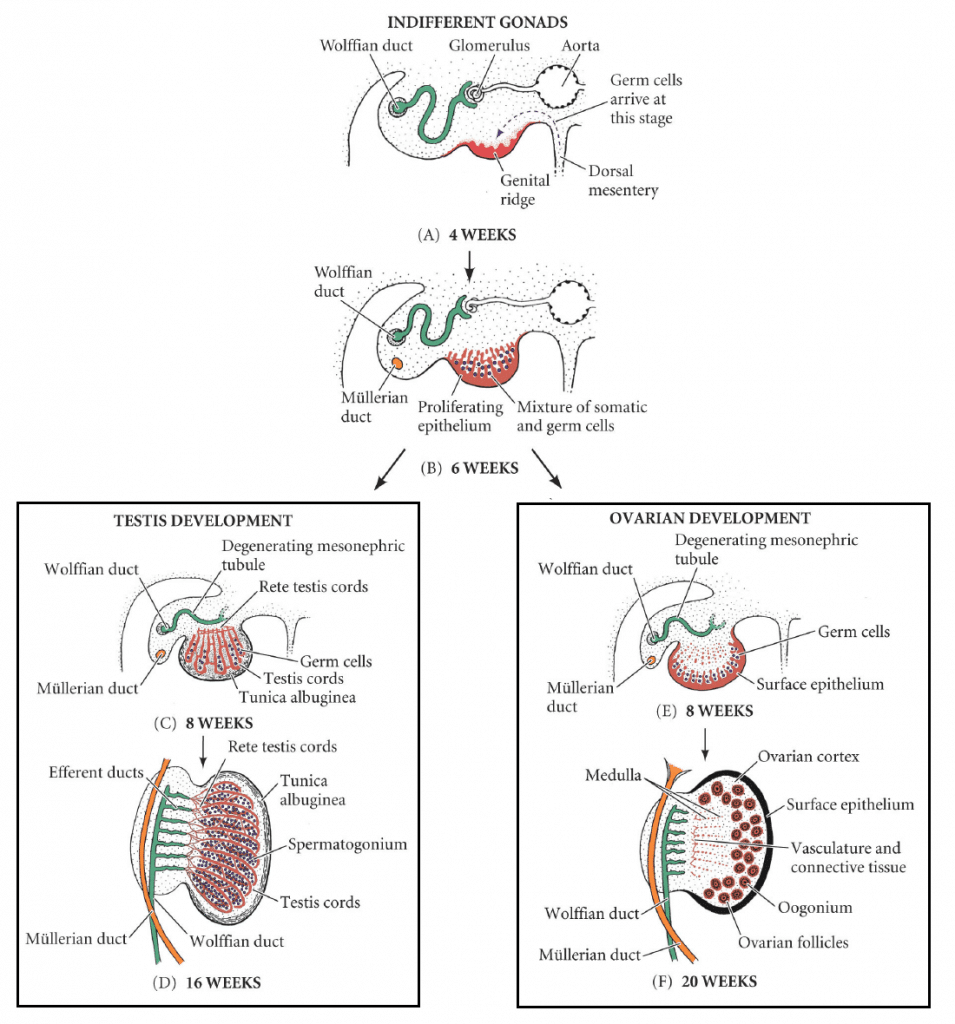
Fig 1
Development of the male and female gonad from the indifferent gonad.
Reference
https://teachmeanatomy.info/the-basics/embryology/reproductive-system/#section-689d9ead508de
Content 2
Content 3
The Gonads
Indifferent Stage
In the first stage of gonadal development, it is impossible to distinguish between the male and female gonad. Thus, it is known as the indifferent stage.
The gonads begin as genital ridges – a pair of longitudinal ridges derived from intermediate mesoderm and overlying epithelium. They initially do not contain any germ cells.
In the fourth week, germ cells begin to migrate from the endoderm lining of the yolk sac to the genital ridges, via the dorsal mesentery of the hindgut. They reach the genital ridges in the sixth week.
Simultaneously, the epithelium of the genital ridges proliferates and penetrates the intermediate mesoderm to form the primitive sex cords. The combination of germ cells and primitive sex cords forms the indifferent gonad – from which development into the testes or ovaries can occur.
Testes
In a male embryo, the XY sex chromosomes are present. The Y chromosome contains the SRY gene, which stimulates the development of the primitive sex cords to form testis (medullary) cords. The tunica albuginea, a fibrous connective tissue layer, forms around the cords.
A portion of the testis cords breaks off to form the future rete testis. The remaining cords contain two types of cells:
- Germ cells
- Sertoli cells (derived from the surface epithelium of the gland).
In puberty, these cords acquire a lumen and become the seminiferous tubules – the site within which sperm will be formed.
Located between the testis cords are the Leydig cells (derived from the intermediate mesoderm). In the eighth week, they begin production of testosterone – which drives differentiation of the internal and external genitalia.
Ovaries
In a female embryo, the XX sex chromosomes are present. As there is no Y chromosome, there is no SRY gene to influence development. Without it, the primitive sex cords degenerate and do not form the testis cords.
Instead, the epithelium of the gonad continues to proliferate, producing cortical cords. In the third month, these cords break up into clusters, surrounding each oogonium (germ cell) with a layer of epithelial follicular cells, forming a primordial follicle.
Fig 1
Development of the male and female gonad from the indifferent gonad.
+++++++++++++++
-
Anatomic differentiation into male or female occurs in utero,
The final maturation of fully functional reproductive organs is not completed until puberty.
-
The complement of sex chromosomes determines sexual differentiation.
-
Female gametes (oocytes) all have a 22X chromosomal makeup, whereas male gametes (spermatozoa) are either 22X or 22Y.
-
The chromosomal sex of the fetus is determined at fertilization when the male and female gametes combine; XX is female and XY is male.
-
-
The default phenotypic sex of the fetus is female if it does not have a Y chromosome.
-
The presence of a Y chromosome directs the undifferentiated gonad to become a testis rather than an ovary.
-
A single gene (SRY), located in the sex-determining region of the Y chromosome, produces testis-determining factor that is required for male sexual differentiation.
-
-
Before sexual differentiation, the fetus has two parallel duct systems located near the undifferentiated gonads: the mesonephric (Wolffian) duct and the paramesonephric (Müllerian) duct
Figure 9-1
Differentiation of the internal reproductive organs. The mesonephric (Wolffian) and paramesonephric (Müllerian) duct systems of the early embryo run lateral to the undifferentiated fetal gonad. Secretion of testosterone in the male fetus results in the development of the mesonephric duct into the male reproductive organs; secretion of Müllerian-inhibiting substance by the Sertoli cells produces regression of the paramesonephric ducts. In the female fetus, the absence of testosterone allows the paramesonephric ducts to develop into the female reproductive organs and results in degeneration of the mesonephric ducts.
-
By week 10 of gestation, the fetal gonads can be distinguished as either testes or ovaries.
-
In males, the primordial germ cells give rise to precursors of male gametes called spermatogonia.
-
The germinal epithelium that will later produce male gametes is formed by spermatogonia plus support cells called Sertoli cells.
-
The surrounding mesenchyme becomes Leydig cells, which secrete testosterone.
-
-
In females, the primordial germ cells give rise to precursors of female gametes called oogonia.
-
The epithelium surrounding the oogonia differentiates into granulosa cells, and the surrounding ovarian mesenchyme becomes thecal cells.
-
In the sexually mature female, estrogens and progestins are secreted by the granulosa and theca cells.
-
-
-
Differentiation of the genitalia depends only on the presence or absence of hormones secreted by the testes.
-
In the male fetus, the secretion of testosterone by Leydig cells directs each mesonephric duct to develop into an epididymis, a vas deferens, and a seminal vesicle.
-
Leydig cells produce testosterone in response to the hormone human chorionic gonadotropin (hCG), which is secreted by the placenta.
-
The developing Sertoli cells are directed by SRY to secrete Müllerian-inhibiting substance, causing regression of the Müllerian duct system.
-
The absence of the Müllerian-inhibiting substance in the female fetus allows the Müllerian duct system (instead of the Wolffian duct) to develop, leading to formation of the fallopian tubes, the uterus, and the upper vagina.
-
Fetal ovaries are not necessary for the development of the female genitalia due to the high concentration of maternal estrogens that are present during pregnancy.
-
-
Undifferentiated external genitalia consist of a genital tubercle and a urogenital slit, bounded by two lateral genital folds and two labioscrotal swellings (Figure 9-2).
-
In males, the conversion of testosterone to dihydrotestosterone, via the enzyme 5α-reductase within these target tissues, is necessary for formation of the prostate gland and the male external genitalia.
-
The genital folds fuse to form the penis; the enlargement and fusion of the labioscrotal swellings form the scrotum.
-
Descent of the fetal testes into the scrotum requires the secretion of the fetal gonadotropins and occurs during the last trimester of pregnancy.
-
 Cryptorchidism is the incomplete descent of the testis from the abdominal cavity to the scrotum and is associated with testicular malignancy and infertility. In the setting of unilateral cryptorchidism, the fully descended testis may remain at risk of impaired sperm production or of becoming malignant.
Cryptorchidism is the incomplete descent of the testis from the abdominal cavity to the scrotum and is associated with testicular malignancy and infertility. In the setting of unilateral cryptorchidism, the fully descended testis may remain at risk of impaired sperm production or of becoming malignant.
-
-
In females, the urogenital slit remains open to form the introitus (vaginal opening).
-
The labia minora are formed from the genital folds, and the clitoris forms anterior to the urethral opening.
-
The labia majora are formed from the labioscrotal swellings. Exposure of the female fetus to androgens at this critical time of sexual differentiation can result in masculinization of the fetus, irrespective of the genetic or gonadal sex.
-
Virilization of a fetus refers to a genetic female with normal ovaries and Müllerian duct structures (e.g., fallopian tubes, uterus, and upper vagina), but with masculinization of the external genitalia (e.g., clitoromegaly, fusion of the labioscrotal folds) due to excessive in utero exposure to androgens.
-
-
-
Differences in sexual development (DSD).
-
There are several conditions in which gonadal and phenotypic sex differ.
-
Complete androgen insensitivity syndrome (CAIS) results from the lack of functional androgen receptors and illustrates the role of steroids in sexual differentiation, as follows:
-
CAIS patients are 46 XY DSD. The gonads become testes since the Y chromosome is present; the testes remain undescended.
-
Müllerian-inhibiting substance continues to be secreted from the Sertoli cells, resulting in the absence of the female internal genitalia. Patients have a short, blind-ended vagina without a cervix, uterus, or ovaries.
-
Dihydrotestosterone is made but cannot direct the Wolffian duct to develop into male genitalia due to lack of androgen receptors; individuals have female external genitalia.
-
Masculinization does not occur during puberty because of the lack of testosterone action. Conversion of testosterone to estrogen causes breast development at puberty instead, but there is only a small amount of pubic hair. Diagnosis is often determined following failure of the onset of the menstrual cycle.
-
-
Deficiency of 5α-reductase is another example of 46 XY DSD and causes ambiguous genitalia because it interferes with the conversion of testosterone to dihydrotestosterone.
-
Testosterone is present but is a weaker androgen than DHT leading to varying degrees of failure of the genital and labioscrotal folds to close.
-
Masculinization at puberty can occur because androgen receptors are present and respond to testosterone, distinguishing this condition from CAIS.
-
-
Congenital adrenal hyperplasia (CAH) is the most common reason for ambiguous genitalia in 46 XX DSD individuals.
-
Deficiency of the enzyme 21-hydroxylase in the steroid synthesis pathway is the most common cause of CAH and results in excessive production of adrenal androgens and virilization of a female fetus.
-
-
-
Puberty (Figure 9-3).
-
Puberty is the final stage in the process of sexual differentiation and results in the mature individual, with the development of physical and behavioral attributes that allow reproduction.
-
Adrenarche is the stage of maturation when the contribution of the adrenal glands occurs before the visible onset of puberty, with increased secretion of adrenal androgens in both males and females.
-
Adrenarche occurs at around 6–8 years of age and is independent of adrenocorticotropic hormone (ACTH) or gonadotropins.
-
Increasing levels of the adrenal androgens initiate axillary and pubic hair growth.
-
-
Puberty begins with activation of the gonadotropin-releasing hormone (GnRH) pulse generator within the hypothalamus.
-
The peptide neurotransmitter Kisspeptin stimulates GnRH neurons.
-
Leptin is among the factors that stimulate Kisspeptin, especially in females, to signal adequate metabolic capacity for reproduction.
-
The early stages of puberty are characterized by an increase in the pulsatile secretion of luteinizing hormone (LH) and follicle-stimulating hormone (FSH), which occurs during sleep.
-
-
In females, visible puberty begins between 8 and 10 years of age, with breast enlargement (thelarche).
-
The first menstrual cycle (menarche) usually occurs between the ages of 11 and 14 years (median age 12.4 years).
-
Female secondary sexual characteristics develop over 2–3 years, mainly in response to increasing ovarian estrogen secretion, and include:
-
Breast development characterized by growth of the lactiferous duct system and deposition of fat. Final differentiation and development of the breast only occurs in pregnancy.
-
Growth of pubic and axillary hair.
-
Enlargement of the uterus, the clitoris, and the labia, and keratinization of the vaginal mucosa.
-
Widening of the pelvis and deposition of fat on the hips and thighs.
-
-
Establishment of the monthly ovarian cycle requires progressive maturation of the hypothalamic-pituitary-ovarian axis:
-
FSH and LH secretion increase.
-
Ovarian steroid secretion occurs in response to FSH and LH.
-
A midcycle positive feedback response to estrogen develops, causing an LH surge that initiates ovulation.
-
Initial menstrual cycles may be anovulatory and are often irregular as the hormonal axis matures.
-
-
Puberty visibly begins in males between 9 and 14 years of age, with enlargement of the testes.
-
The first ejaculation (thorarche) occurs between the ages of 12 and 14 years.
-
The testes increase in size and undergo maturation, involving secretion of testosterone by Leydig cells and spermatogenesis in the seminiferous tubules.
-
Enlargement of the testes in puberty results from FSH secretion and can be used as a clinical indicator of normal function in the developing hypothalamic-pituitary axis.
-
-
The scrotum and penis enlarge.
-
Secondary sexual characteristics develop with increasing testosterone levels and include increases in:
-
Hair on the axilla, face, trunk, and pubis.
-
Bone mass.
-
Mass and strength of skeletal muscle.
-
Size of the larynx with associated deepening of the voice.
-
-
-
Pubertal growth spurt.
-
Somatic growth is primarily controlled by the growth hormone (GH)–insulin-like growth factor-1 (IGF-1) endocrine axis.
-
Increased LH and FSH secretion at the beginning of puberty occurs together with increased GH secretion.
-
The increase in GH secretion is caused by increasing estrogen levels in both sexes; in males, estrogen is produced from testosterone via the enzyme aromatase.
-
The highest rates of GH secretion occur during puberty because the strength of negative feedback inhibition of GH secretion by IGF-1 is weak. After puberty, the rate of GH production declines with age.
-
-
Linear growth is completed in females by about age 17 and in males by about age 21. On average, men are 10–15 cm taller than women because:
-
Growth during the pubertal growth spurt is more rapid in males than in females.
-
Puberty occurs later in males, allowing about 2 additional years of prepubertal growth before closure of the epiphyseal growth plates in long bones, which occurs in response to sex steroids.
-
-
Precocious puberty refers to early pubertal development, often before 8 years of age in girls and before 9 years of age in boys. In addition to early sexual maturation there is increased linear growth and skeletal maturation. Children who experience precocious puberty are often tallfor their age during childhood, but short for their age during adulthood.
-
Several genetic abnormalities cause delayed or absent puberty:
-
Females with Turner syndrome (45 XO) lack one of their X-chromosomes and have rudimentary “streak” ovaries. There is phenotypic female sexual differentiation in utero but the inability to undergo puberty without hormone therapy.
-
Males with Kleinfelter syndrome (47 XXY) have an additional X chromosome, resulting in small testes and low testosterone levels. Puberty is often delayed or incomplete.
-
Kallmann syndrome is a gene (not chromosomal) defect, which leads to lack of GnRH neurons. The condition is more common in males due to X-linked inheritance of one of the causative genes. The syndrome causes hypogonadotropic hypogonadism. Patients with Kallmann syndrome also have decreased or absent sense of smell (anosmia).
-
-

Figure 9-2
Differentiation of the external genitalia. A. Structures of the undifferentiated external genitalia. B. In the male fetus, fusion of the genital folds creates the penis, and fusion of the labioscrotal swellings forms the scrotum. C. In the female fetus, the genital folds do not fuse, allowing the vagina and urethra to open between the labia minora; the labia majora are formed from the labioscrotal swellings.


{kind=link}